L’intolleranza ortostatica è un argomento confuso. Parte della confusione proviene dal recente apprezzamento delle varianti cliniche della condizione, alcuni proviene dalla comprensione emergente delle sue diverse fisiopatologie sottostanti e alcuni proviene dalla sua nomenclatura, che sembra cambiare almeno ogni anno.
Il termine ortostasi significa letteralmente in piedi. L’intolleranza ortostatica (OI) può essere definita come “lo sviluppo di sintomi in posizione eretta, durante la posizione eretta che sono alleviati dalla posizione sdraiata.”Sebbene l’uso di un termine come intolleranza ortostatica implichi logicamente la presenza di segni e sintomi in posizione verticale, le variazioni nel flusso sanguigno e nella regolazione della pressione sanguigna (BP) si trovano anche in posizione supina o seduta, ma possono richiedere attrezzature speciali per rilevare e quindi non possono essere facilmente distinguibili fino a quando lo stress ortostatico diventa evidente.
Stare in piedi con successo richiede l’interazione di volume del sangue, fattori fisici, neurologici, umorali e vascolari che compensano gli effetti del pooling venoso gravitazionale. In condizioni ordinarie, le alterazioni umorali acute hanno poco a che fare con la risposta iniziale a stare in piedi, ma possono svolgere un ruolo importante durante l’intolleranza ortostatica cronica o relativamente tardi durante la posizione eretta. Inoltre, i cambiamenti in tali fattori possono influenzare le risposte a riposo o toniche e quindi possono influenzare la regolazione vascolare generale attraverso effetti di fondo.
Se i sintomi iniziano in posizione supina, allora non c’è OI. L’OI transitoria si verifica comunemente durante la disidratazione o la malattia infettiva. Segni e sintomi tipici includono: perdita di coscienza o deficit cognitivi minori (perdita di memoria, diminuzione del ragionamento e della concentrazione); difficoltà visive; stordimento; mal di testa; affaticamento; aumenti di BP (ipertensione), diminuzioni di BP (ipotensione); debolezza; nausea e dolore addominale; sudorazione; tremulousness; e intolleranza all’esercizio. A meno che in pericolo (ad esempio, in piedi su una scogliera), OI non è letale. Alcuni risultati OI, come nausea e sudorazione riguardano direttamente l’attivazione autonomica. Tuttavia, perdita di coscienza, grave stordimento e perdita neurocognitiva si riferiscono alla disfunzione del sistema nervoso centrale (SNC) e obbligano la reclinazione.
I sintomi del SNC sono prodotti dal flusso sanguigno cerebrale alterato che coinvolge forse il tronco cerebrale. La velocità del flusso sanguigno cerebrale (CBF) è mostrata nell’immagine sottostante per due forme comuni di OI, sincope vasovagale (VVS) e sindrome da tachicardia posturale (POTS). L’autoregolazione cerebrale può essere compromessa come in POTS e VVS e può essere ridotta dall’iperventilazione e dalla vasocostrizione cerebrale ipocapneica. L’iperventilazione posturale involontaria, per lo più iperpnea, è osservata in tutti i pazienti VVS e nel 50% dei pazienti POTS nel nostro laboratorio. L’attività del nervo trigemino, simpatico o parasimpatico può anche influenzare il CBF ortostatico.
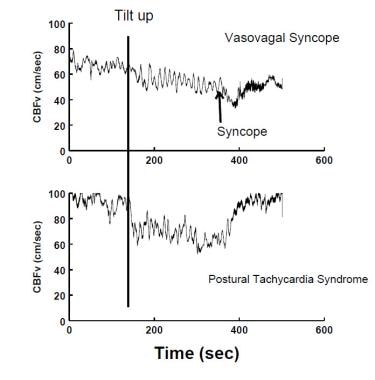 Diminuzione del CBFv misurata mediante ecografia Doppler transcranica si verifica durante un VVS (pannello superiore) e nella sindrome da tachicardia posturale (POTS) (pannello inferiore). Durante VVS, CBF diminuisce gradualmente in un primo momento e poi più bruscamente come il paziente acutamente perde conoscenza. Nei vasi, il CBF è ridotto in modo abbastanza uniforme; non c’è perdita di coscienza sebbene la vertigini sia tipica.
Diminuzione del CBFv misurata mediante ecografia Doppler transcranica si verifica durante un VVS (pannello superiore) e nella sindrome da tachicardia posturale (POTS) (pannello inferiore). Durante VVS, CBF diminuisce gradualmente in un primo momento e poi più bruscamente come il paziente acutamente perde conoscenza. Nei vasi, il CBF è ridotto in modo abbastanza uniforme; non c’è perdita di coscienza sebbene la vertigini sia tipica. L’intolleranza ortostatica non è sempre dovuta a disfunzione autonomica o di altra natura compensatoria e può essere dovuta a risposte inadeguate dei meccanismi compensatori ai fattori di stress ambientali. Ad esempio, chi è disidratato può non essere in grado di alzarsi in piedi senza conseguente ipotensione ortostatica, ma non è presente alcuna disfunzione autonomica e quindi non è presente ipotensione ortostatica neurogena; invece, i sistemi regolatori autonomi e altri non possono compensare adeguatamente la perdita di volume del sangue circolante.
D’altra parte, l’OI neurogenica, in cui la vasocostrizione adrenergica è difettosa, è associata a insufficienza autonomica primaria; i pazienti con malattie correlate non possono rimanere in piedi e hanno anomalie autonomiche rilevabili in tutte le posizioni posturali. Pertanto, OI comprende qualsiasi condizione con flusso sanguigno, frequenza cardiaca e inadeguatezza della regolazione cardiorespiratoria dimostrabile in posizione verticale ma che può anche avere risultati anormali in tutte le posizioni. In tali circostanze, l’OI è spesso la manifestazione più ovvia di una compromissione più diffusa nella fisiologia neurovascolare integrativa.
I vari tipi di OI sono discussi nelle sezioni che seguono.
Ipotensione ortostatica iniziale
L’ipotensione ortostatica (OH) è definita come una riduzione sostenuta della pressione sistolica>20 mmHg o pressione diastolica>10 mmHg entro 3 minuti dalla posizione eretta o dopo l’inclinazione head-up a ≥60o. Il requisito di una riduzione sostenuta esclude l’ipotensione ortostatica iniziale (IOH). Questa definizione è relativamente recente ed è stata assemblata da un gruppo di consenso nel 2011. Prima di allora, non c’era una definizione coerente di OH. L’OH non neurogeno può essere causato da farmaci, disidratazione, perdita di sangue, età e malattie che causano secondariamente ipovolemia acuta o cronica. Neurogena OH è identificato con insufficienza autonomica a causa di insufficiente rilascio di noradrenalina da neuroni vasomotori simpatici che porta al fallimento vasocostrittore. L’OH neurogenico è raro nei giovani poiché la maggior parte delle cause di insufficienza autonomica sono acquisite con l’età sia come primaria (ad es., insufficienza autonomica pura, PAF) o malattia secondaria (diabete). Il fallimento autonomo può essere primario con forme pre-gangliari, post-gangliari o entrambe(ad esempio, malattia di Parkinson) di disfunzione simpatica. Tuttavia, esistono varianti genetiche congenite come la Disautonomia familiare (sindrome di Riley-Day) e la carenza di beta-idrossilasi della dopamina squisitamente rara (carenza di DBH). Il fallimento autonomo può essere autoimmune e può presentarsi con la sindrome post-infettiva di Guillain-Barre anche se la disfunzione autonomica sembra avere scarso effetto sul risultato finale. Il fallimento autonomo è più comunemente acquisito come un aspetto secondario della malattia sistemica come il diabete. La denervazione cardiaca simpatica è un aspetto centrale della malattia di Parkinson e può essere trovata in altre forme di insufficienza autonomica. Anche l’innervazione parasimpatica cardiaca è spesso difettosa con conseguente calo costante della PA con tachicardia riflessa ridotta durante la sfida ortostatica come mostrato nell’immagine qui sotto.
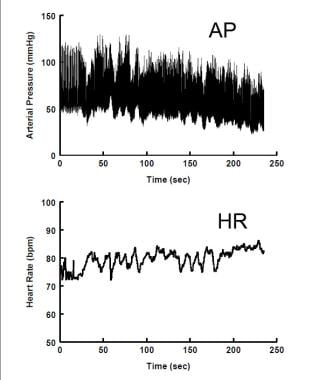 Ipotensione ortostatica neurogena. La pressione arteriosa (pannello superiore) diminuisce costantemente durante la posizione verticale, mentre la frequenza cardiaca (pannello inferiore) è solo leggermente aumentata.
Ipotensione ortostatica neurogena. La pressione arteriosa (pannello superiore) diminuisce costantemente durante la posizione verticale, mentre la frequenza cardiaca (pannello inferiore) è solo leggermente aumentata. L’OH non neurogeno è relativamente comune nei giovani. Può essere causato da farmaci o ipovolemia (ad esempio, disidratazione, emorragia). È di gran lunga la forma più comune di OH nei giovani. Non vi è alcun fallimento della funzione autonomica, ma piuttosto una compensazione ANS incompleta per fattori di stress non autonomi eccessivi.
Neurogena OH (NOH) significa grave malattia autonomica. È identificato con vero fallimento vasocostrittore autonomo a causa del rilascio inadeguato di noradrenalina dai nervi simpatici e HR non può aumentare in modo appropriato con in piedi.
Sindrome da tachicardia posturale
La sindrome da tachicardia posturale (POTS) è definita dai sintomi quotidiani di intolleranza ortostatica (OI) associati a tachicardia verticale eccessiva ma non a ipotensione (vedere immagine sotto).
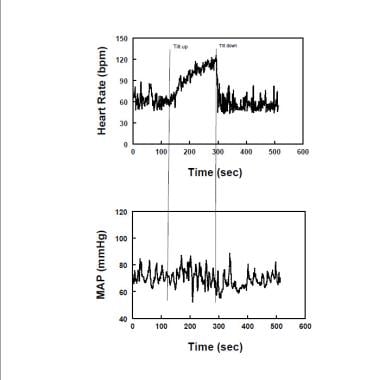 Diagramma che mostra la frequenza cardiaca rappresentativa (pannello superiore) e la pressione arteriosa media (MAP-pannello inferiore) durante l’inclinazione verticale in un paziente con sindrome da tachicardia posturale (POTS). La frequenza cardiaca aumenta, mentre la MAPPA è stabile per tutta l’inclinazione in VASO.
Diagramma che mostra la frequenza cardiaca rappresentativa (pannello superiore) e la pressione arteriosa media (MAP-pannello inferiore) durante l’inclinazione verticale in un paziente con sindrome da tachicardia posturale (POTS). La frequenza cardiaca aumenta, mentre la MAPPA è stabile per tutta l’inclinazione in VASO. L’eccessiva tachicardia negli adulti è definita da un aumento verticale dell’HR superiore a 30 bpm o da una frequenza cardiaca superiore a 120 bpm. Ricordiamo che la normale risposta HR all’ortostasi è un aumento della HR mentre i pazienti con insufficienza autonomica spesso non hanno un aumento significativo della HR in posizione verticale. Incrementi di frequenza cardiaca più grandi sono osservati nei giovani con vasi, che è importante sapere per evitare la sovra-diagnosi. I sintomi di OI devono essere concomitanti con l’eccessiva tachicardia. Nessun sintomo, nessun VASO. Tachicardia e sintomi concomitanti sono molto spesso osservati durante test ortostatici estremamente prolungati che devono quindi essere evitati se si deve effettuare la diagnosi specifica di POTS. POTS è stato spesso diviso in sottogruppi designati “POT neuropatici”, in cui si presume che parziale denervazione simpatica o ipoattività adrenergica è presente, e” POT iperadrenergici”, in cui sovrattività adrenergica verticale domina l’immagine.
VASI neuropatici
Come originariamente descritto, i VASI neuropatici sono causati da una diminuzione della vasocostrizione simpatica adrenergica negli arti inferiori, associata a una ridotta fuoriuscita di noradrenalina alle gambe e a una ridotta vasocostrizione degli arti inferiori. C’è spesso un aumento del flusso sanguigno (“alto flusso”) negli arti inferiori anche in posizione supina. Un’altra variante neuropatica ha l’emodinamica normale degli arti inferiori (“flusso normale”) ma ha diminuito la vasocostrizione adrenergica simpatica regionale nella circolazione splancnica. I VASI neuropatici possono rappresentare una neuropatia autonomica autoimmune. Così, quando in posizione verticale, vasi neuropatici pazienti hanno maggiore del normale ridistribuzione del sangue alla vascolarizzazione dipendente causando baroreflex mediata tachicardia e vasocostrizione. La risposta cardiaca baroreflex è anche smussata in vasi. L’ipovolemia centrale può anche provocare iperpnea e ipocapnia in quasi il 50% dei pazienti attraverso un meccanismo mediato da baroreflex.
VASI iperadrenergici
La tachicardia dei VASI iperadrenergici è causata da un aumento del potenziamento adrenergico pre-sinaptico o post-sinaptico. Ciò potrebbe includere attività simpatica centrale e aumento dell’attività del nervo simpatico. È stato riportato un aumento dell’attività simpatica supina, ma non universalmente. Ad oggi, il nostro laboratorio ha osservato solo un aumento dell’attività simpatica muscolare verticale in un paziente POTS. Una causa di VASI iperadrenergici è l’aumento della noradrenalina sinaptica. L’eterozigote di carenza del trasportatore della noradrenalina è il primo esempio di questo meccanismo ma è stato trovato come mutazione autosomica in soltanto un pedigree. La carenza meno severa, possibilmente epigenetica del trasportatore della norepinefrina (RETE) inoltre è stata dimostrata recentemente e può avere una più ampia prevalenza.
Considerazioni alternative sul meccanismo includono la modulazione della sinapsi adrenergica attraverso una maggiore sintesi e rilascio di noradrenalina e una maggiore affinità post-sinaptica, che può essere modulata da trasmettitori locali e umorali. Così, per esempio, le azioni reciproche di ossido nitrico (NO) e angiotensina-II rispettivamente riducono e migliorano l’attività adrenergica. Il ruolo di NO come neurotrasmettitore inibitorio è ora ben noto. Nitrergico NO rilasciato dai nervi che hanno attività parasimpatica agire a siti pre-sinaptici e post-sinaptici per diminuire la trasduzione adrenergica, il processo mediante il quale un impulso nervoso simpatico provoca la contrazione della muscolatura liscia vascolare. Ciò include la riduzione del rilascio e del legame della noradrenalina dalla sinapsi neurovascolare, l’interferenza con la neurotrasmissione post-sinaptica, la denaturazione chimica della noradrenalina e la down-regolazione dei recettori adrenergici.
Al contrario, gli studi di simpatoeccitazione mostrano che l’angiotensina-II agisce attraverso i recettori AT1 per aumentare la produzione di ossigeno reattivo (ROS) e specie di azoto all’interno del cervello a neuroni simpatici pre-sinaptici e agiscono nella periferia dove producono aumento pre – e post-sinaptico della trasduzione simpatica e upregulation dei recettori adrenergici. Inoltre, il rilascio e il legame della noradrenalina sono facilitati, così come gli effetti della noradrenalina, in presenza di angiotensina-II. Ciò dipende criticamente dalla formazione di ROS, che diminuisce anche NO, spesso disaccoppiando l’ossido nitrico sintasi. Questo meccanismo si verifica in una variante di “VASI iperadrenergici” associati a tachicardia, pallore, vasocostrizione (“basso flusso”) e ipovolemia assoluta anche in posizione supina. NO, la renina plasmatica e l’aldosterone sierico sono diminuiti, mentre l’angiotensina-II plasmatica è aumentata da un difetto nell’ACE-2.
Sincope posturale (sincope vasovagale, acuta OI, semplice svenimento)
La sincope (svenimento) è definita come “completa perdita di coscienza dovuta a ipoperfusione cerebrale globale transitoria caratterizzata da insorgenza rapida, breve durata e recupero completo spontaneo.”La maggior parte della sincope è causata da ipotensione sistemica e riduzione del flusso sanguigno cerebrale. È possibile che un incidente cerebrovascolare potrebbe presentare in modo simile, anche se non è mai stato segnalato in pediatria. La sincope può essere causata da ipotensione ortostatica (OH), che è già stata discussa. OH è facilmente escluso da un test in piedi di 3 minuti (vedi la figura sotto).
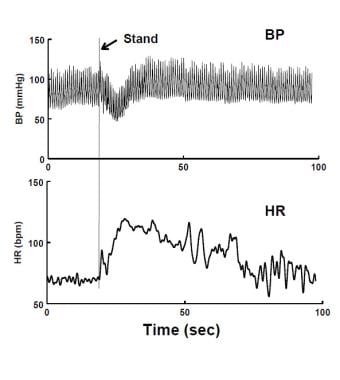 Ipotensione ortostatica immediata (IOH) in posizione eretta. C’è una diminuzione di breve durata della pressione sanguigna (BP – pannello superiore) e aumento della frequenza cardiaca (HR – pannello inferiore). La caduta della pressione arteriosa viene risolta entro 20 secondi. Il paziente ha avuto stordimento transitorio.
Ipotensione ortostatica immediata (IOH) in posizione eretta. C’è una diminuzione di breve durata della pressione sanguigna (BP – pannello superiore) e aumento della frequenza cardiaca (HR – pannello inferiore). La caduta della pressione arteriosa viene risolta entro 20 secondi. Il paziente ha avuto stordimento transitorio. La sincope è divisa tra sincope cardiovascolare, malattia cardiopolmonare frequentemente aritmica o strutturale e sincope riflessa o neuralmente mediata. La sincope cardiogenica può essere pericolosa per la vita e ha una prognosi infausta a meno che non venga trattata la fisiopatologia cardiaca. La sincope cardiogenica non è OI perché la recumbency non produce specificamente un miglioramento. La sincope riflessa ha una buona prognosi. La sincope riflessa include sincope vasovagale e sincope situazionale inclusa la sincope del seno carotideo, che è essenzialmente sconosciuta alla pediatria. Deglutition, defecazione, minzione, e sincope tosse sono raramente osservati nei giovani; e governare i capelli e adolescente tratto sincope varianti sono particolari per l’adolescenza. Lo svenimento durante l’esercizio solleva una “bandiera rossa” per la sincope cardiogenica e l’ulteriore attività sportiva dovrebbe essere ridotta fino al completamento della valutazione cardiaca. Tuttavia, la causa più comune di sincope legata all’esercizio nei giovani isVVS. La sincope cardiogenica, anche se di solito non è correlata posturalmente, non può essere automaticamente respinta dopo un primo svenimento. Pertanto, i primi e conseguenti episodi prima della valutazione cardiovascolare devono essere trattati come urgenti. Se lo svenimento viene successivamente trovato non cardiogeno, l’urgenza viene ridotta e le semplici manovre spesso sono sufficienti per affrontare acutamente le circostanze. I cardiologi sono spesso coinvolti nella valutazione precoce della sincope perché le valutazioni iniziali dovrebbero determinare se la condizione è di eziologia cardiaca o non cardiaca. Le malattie cardiache, una volta trovate, sono trattate in modo specifico. La sincope cardiaca può manifestarsi prima durante l’esercizio, il che pone lo stress più fisiologico sulle circolazioni coronariche, sistemiche e polmonari e sulla funzione cardiaca complessiva. Sincope correlata all’esercizio fisico o sincope con sintomi cardiaci (ad esempio, tachicardia, dolore toracico) indica una ricerca di malattie cardiache sottostanti. Tuttavia, la sincope legata all’esercizio nei giovani è più spesso non cardiogena e la fisiologia può assomigliare a un semplice svenimento.
Tuttavia, anche quando è coinvolta l’ipotensione neurocardiogenica, la fisiopatologia e la storia clinica possono essere più complesse, coinvolgendo cambiamenti nella respirazione con dispnea e disfunzione chemoreflex e baroreflex che potrebbero suggerire malattie cardiache provocate dall’esercizio. Tuttavia, nonostante il relativo grado di preoccupazione relativo allo svenimento nel giovane atleta, la maggior parte di questi episodi sono di origine non cardiogena. Tuttavia, ciò non dovrebbe ovviare alla necessità di valutare tali pazienti per malattie cardiache potenzialmente letali. La sincope cardiogenica è ben descritta altrove e non è discussa qui.
La sincope posturale e la sincope emotiva o fobica comprendono VVS, il più grande sottogruppo all’interno della categoria della sincope riflessa. La perdita di vasodilatazione a livello regionale o di sistema è un elemento in tutti i VV, almeno come evento terminale; potrebbe non essere sempre dovuto alla perdita dell’attività del nervo simpatico. La sincope posturale è acuta EI e circa due terzi dei pazienti sono di sesso femminile, mentre gli adolescenti con questo tendono ad essere alti, magri e in rapida crescita. La perdita di coscienza è spesso preceduta da un prodromo di sintomi OI, in particolare stordimento, nausea, sudorazione, debolezza e visione offuscata. Tradizionalmente, si credeva che la sincope posturale fosse dovuta ai riflessi di un cuore ipercontrattile e sottopieno analogo al riflesso di Bezold-Jarisch. La prova del contrario è maturata; tale stimolo sarebbe di breve durata. A causa dello scarico del barocettore, pochissimi nervi afferenti erano eccitati negli esperimenti originali di Oberg e Thoren nel gatto moribondo con emorragia. VVS può verificarsi inun ricevente di trapianto denervato ventricolare e il cuore non è né vuoto né ipercontrattile prima della sincope. Ancora non comprendiamo completamente la fisiopatologia del semplice debole.
Nella variante più comune del debole posturale che si verifica nei pazienti giovani, il debole posturale comprende tre fasi (vedi l’immagine sotto), che assomigliano fortemente ai cambiamenti circolatori riscontrati durante l’emorragia.
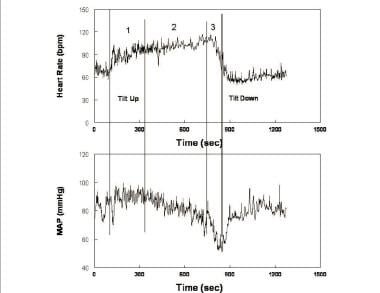 Frequenza cardiaca (pannello superiore) e pressione arteriosa media (pannello MAP – inferiore) durante l’inclinazione verticale in un paziente sincope posturale rappresentativo. I cambiamenti durante l’inclinazione si verificano in tre fasi: durante il primo stadio (1), dopo l’ipotensione iniziale, MAP si stabilizza leggermente più in alto della pressione a riposo mentre la frequenza cardiaca aumenta. Durante la seconda fase (2), la MAPPA inizia a diminuire gradualmente, mentre la frequenza cardiaca continua ad aumentare. Si noti che l’incremento della frequenza cardiaca da supina a verticale soddisfa i criteri di tachicardia per la sindrome da tachicardia posturale (POTS). Durante la terza fase (3), MAP e poi la frequenza cardiaca cadono bruscamente e rapidamente come la perdita di coscienza sopravviene.
Frequenza cardiaca (pannello superiore) e pressione arteriosa media (pannello MAP – inferiore) durante l’inclinazione verticale in un paziente sincope posturale rappresentativo. I cambiamenti durante l’inclinazione si verificano in tre fasi: durante il primo stadio (1), dopo l’ipotensione iniziale, MAP si stabilizza leggermente più in alto della pressione a riposo mentre la frequenza cardiaca aumenta. Durante la seconda fase (2), la MAPPA inizia a diminuire gradualmente, mentre la frequenza cardiaca continua ad aumentare. Si noti che l’incremento della frequenza cardiaca da supina a verticale soddisfa i criteri di tachicardia per la sindrome da tachicardia posturale (POTS). Durante la terza fase (3), MAP e poi la frequenza cardiaca cadono bruscamente e rapidamente come la perdita di coscienza sopravviene. Dopo l’ipotensione ortostatica iniziale e il ripristino dell’omeostasi circolatoria, la BP si stabilizza mentre l’HR aumenta nello stadio 1. BP stabilità distingue posturale debole dal vero OH. BP presenta spesso fluttuazioni ritmiche durante questa fase denominata “onde di Mayer” con un periodo approssimativo di 10 secondi (0,1 Hz). Una periodicità simile è condivisa dalle fluttuazioni della frequenza cardiaca, dell’attività del nervo simpatico e della resistenza periferica. Le fluttuazioni sono il tempo a ciclo chiuso per la risposta simpatica del baroreflex (cioè il tempo necessario per il rilevamento e la compensazione delle variazioni della BP). Le oscillazioni sono accentuate durante le riduzioni centrali del volume del sangue come si verificano durante l’ortostasi. Durante questa fase la resistenza periferica totale aumenta per sostenere la BP di fronte a una ridotta gittata cardiaca (vedere l’immagine sotto).
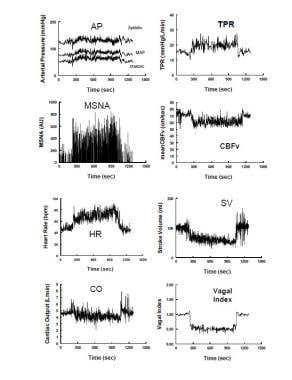 Alterazioni emodinamiche e neurovascolari durante l’inclinazione verticale in un volontario sano rappresentativo. Il pannello sinistro mostra dall’alto verso il basso: pressione arteriosa, attività del nervo simpatico muscolare (MSNA) dal nervo peroneo, frequenza cardiaca (HR) e gittata cardiaca. Il pannello di destra mostra dall’alto verso il basso: resistenza periferica totale (TPR), velocità del flusso sanguigno cerebrale (CBFv) mediante ecografia Doppler transcranica, volume della corsa e un indice vagale calcolato dalla componente aritmia sinusale respiratoria dello spettro di frequenza della variabilità HR. Durante l’inclinazione verticale a 275 secondi, la pressione sistolica, diastolica e arteriosa aumenta leggermente, mentre la pressione del polso diminuisce con una diminuzione del volume della corsa di circa il 40%. HR aumenta in modo che la gittata cardiaca è diminuita solo del 20% a causa dell’aumento di HR. CBFv diminuisce del 5-10%. Sia la resistenza vascolare periferica totale che l’attività del nervo simpatico muscolare aumentano, mentre l’indice vagale diminuisce, riflettendo, rispettivamente, l’attivazione simpatica e il ritiro parasimpatico.
Alterazioni emodinamiche e neurovascolari durante l’inclinazione verticale in un volontario sano rappresentativo. Il pannello sinistro mostra dall’alto verso il basso: pressione arteriosa, attività del nervo simpatico muscolare (MSNA) dal nervo peroneo, frequenza cardiaca (HR) e gittata cardiaca. Il pannello di destra mostra dall’alto verso il basso: resistenza periferica totale (TPR), velocità del flusso sanguigno cerebrale (CBFv) mediante ecografia Doppler transcranica, volume della corsa e un indice vagale calcolato dalla componente aritmia sinusale respiratoria dello spettro di frequenza della variabilità HR. Durante l’inclinazione verticale a 275 secondi, la pressione sistolica, diastolica e arteriosa aumenta leggermente, mentre la pressione del polso diminuisce con una diminuzione del volume della corsa di circa il 40%. HR aumenta in modo che la gittata cardiaca è diminuita solo del 20% a causa dell’aumento di HR. CBFv diminuisce del 5-10%. Sia la resistenza vascolare periferica totale che l’attività del nervo simpatico muscolare aumentano, mentre l’indice vagale diminuisce, riflettendo, rispettivamente, l’attivazione simpatica e il ritiro parasimpatico. Durante la fase 2, la BP diminuisce lentamente man mano che il baroreflex aumenta ulteriormente l’HR. La diminuzione della PA è più spesso correlata alla diminuzione della gittata cardiaca, anche se l’attività simpatica e la resistenza arteriosa periferica sono sostenute. Successivamente, le oscillazioni di resistenza e pressione diminuiscono nonostante la simpatoeccitazione sostenuta. Iperpnea e ipocapnia si verificano in questa fase nella maggior parte dei pazienti. In alcuni pazienti la fase 2 è abbreviata. Ciò è particolarmente vero per i pazienti con sincope convulsiva in cui gli episodi si verificano bruscamente in associazione con asistolia.
La sincope convulsiva o asistolica (vedi l’immagine sotto) si distingue dall’epilessia per la diminuzione dell’attività EEG nella prima e per la risoluzione quasi immediata della postura opistotonica per reclinazione. Nonostante le apparenze, gli svenimenti asistolici non sono cardiogeni ma mediati dal riflesso e sono una forma relativamente rara di svenimento vasovagale semplice che può anche essere trovato nello svenimento fobico.
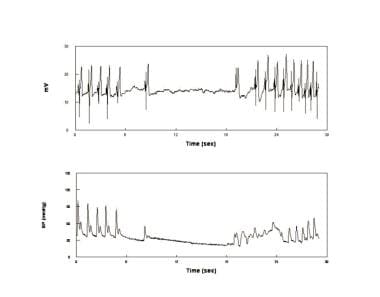 Un debole asistolico. Elettrocardiogramma (pannello superiore) e pressione sanguigna (BP) (pannello inferiore) in un paziente che ha avuto un’asistolia durante lo svenimento. Questo è episodico, relativamente raro e non correlato alla malattia intrinseca del nodo del seno. Gli svenimenti asistolici sono associati alla postura opistotonica e sono stati talvolta indicati come sincope convulsa.
Un debole asistolico. Elettrocardiogramma (pannello superiore) e pressione sanguigna (BP) (pannello inferiore) in un paziente che ha avuto un’asistolia durante lo svenimento. Questo è episodico, relativamente raro e non correlato alla malattia intrinseca del nodo del seno. Gli svenimenti asistolici sono associati alla postura opistotonica e sono stati talvolta indicati come sincope convulsa. In alcuni pazienti sono stati proposti diversi meccanismi per la VVS. I pazienti con VVS con diminuzione della PA a riposo avevano ridotto la tirosina idrossilasi e la sintesi di NE e un gruppo di normotesi aveva una RETE in eccesso. È stato anche dimostrato un deficit selettivo di vasocostrizione splancnica e venocostrizione. I sintomi prodromici OI iniziano durante la fase 2 e i medici potrebbero quindi intrattenere una diagnosi di vasi in ambiente di laboratorio. Storia clinica offre il modo migliore per distinguere i pazienti con acuta episodica sviene con lunghi periodi privi di sintomi (posturale, sincope), da PENTOLE, in cui i sintomi sono cronicamente presente; Infatti, il prodromo di un semplice svenimento e segni e sintomi di PENTOLE sono simili in quanto possono avere simili iniziale fisiopatologia – eccessiva riduzione del volume sanguigno centrale con conseguente tachicardia riflessa. Gli svenimenti posturali, corrispondenti a pazienti con VASI iperadrenergici pallidi e vasocostrittori, non sono generalmente osservati. Per la maggior parte, nella nostra esperienza, i pazienti POT hanno sintomi quotidiani ma non svengono, mentre i pazienti sincope hanno svenimenti episodici ma non sintomi quotidiani. Questa distinzione è diventata meno chiara con il tempo. Pertanto, alcuni pazienti OI cronici (POTS) svengono e alcuni svenimenti episodici hanno anche sintomi quotidiani sottostanti di OI. Tuttavia, svenimento dei pazienti POTS in laboratorio deve essere visto con cautela e non può, di per sé, essere considerato come prova di svenimento del mondo reale. Una storia clinica” reale ” compatibile con lo svenimento è obbligatoria.
Nell’ultima fase, Fase 3, CBF, BP e HR cadono rapidamente in quell’ordine, apparentemente sfidando la causalità BP–CBF. Effetti simili sono spesso visti in sistemi non lineari di tutti i tipi ogni volta che un segnale esterno sufficientemente forte trascina segnali collegati. Pertanto, lavori recenti mostrano che sia le braccia efferenti cardiovagali che simpatiche baroreflex sono compromesse prima dello svenimento e le onde di Mayer scompaiono. Allo stesso modo autoregolazione cerebrale diventa compromessa con trascinamento di CBF, BP, e HR da un segnale estrinseco, che può essere la respirazione iperpneica. Perché l’integrità baroreflex è perso non è ancora noto, ma questo si traduce in anormale BP-HR e BP-MSNA relazioni funzionali tali che HR, BP, e l’attività del nervo simpatico tutti diminuiscono con conseguente bradicardia, ipotensione, e il silenzio simpatico. Il debole è associato a una marcata vasodilatazione sistemica mentre CBF cade con BP in declino. Il lavoro recente sfida la necessità del silenzio comprensivo come precipitante dell’ipotensione finale. Mentre la vasodilatazione si verifica sempre, il baroriflesso simpatico può fallire con o senza silenzio simpatico a causa di una perdita della relazione funzionale tra pressione sanguigna e attivazione simpatica. La perdita di connessioni funzionali tra BP e attività del nervo simpatico, ma non la frequenza cardiaca, si verifica in pazienti con sincope vasodepressiva in cui si verifica vasodilatazione senza bradicardia. Mentre c’è una perdita del baroreflex efferente simpatico che causa la perdita progressiva di vasocostrizione compensatoria, il baroreflex cardiovagal rimane intatto.
I vasi e la sincope posturale sono entrambi associati all’iperventilazione iperpneica. L’iperpnea e l’ipocapnia risultante precedono l’incoscienza praticamente in ogni paziente sincope vasovagale. L’ipotensione e la bradicardia potrebbero essere spiegate dal riflesso di stiramento polmonare senza restrizioni da effetti baroriflessi compensatori. La causa dell’iperpnea non è chiara, ma può riguardare il braccio efferente ventilatorio del baroriflesso arterioso. Risultati simili di iperpnea si trovano in circa il 50% dei pazienti POTS con ipovolemia centrale che non svengono.
La sincope molto frequente o estremamente prolungata può indicare sincope psicogena o risposte di conversione. Questi sono facilmente distinti dalla sincope vera in laboratorio perché non c’è ipotensione o CBF ridotto, ma gli attacchi possono essere reali per il paziente. Alcuni pazienti possono aver avuto VVS bonafide intervallati da episodi psicogeni più frequenti come risposte apprese o condizionate. Una scuola di pensiero suggerisce che tali pazienti sperimentano effettivamente i sintomi del vero VVS senza i segni.
L’intolleranza ortostatica è comune ma spesso fraintesa. L’indagine della circostanza è un campo in evoluzione dello studio fisiologico integrativo. VVS posturale è identificato con intolleranza ortostatica acuta. Nonostante la sua ubiquità, gli scienziati non capiscono ancora perché alcune persone svengono. POTS è identificato con intolleranza ortostatica cronica. POTS, tuttavia, rimane un’entità eterogenea, probabilmente di varie eziologie. Fino a quando non si ottiene una migliore comprensione, il trattamento rimane più congetture della scienza.