Um sistema Baseado Revisão do Papel do Oftalmologista no Diagnóstico da Doença de Wilson
Emily S. Birkholz, MD, Thomas A. Oetting, MS, MD
em 28 de julho de 2009
Queixa principal: Referência para a avaliação de Kayser-Fleischer Anéis
a História do Presente Doença: O paciente era um homem de 25 anos de idade que tinha experimentado uma ternura subcostal direita e aumento da fadiga nos últimos meses. Ele também tinha notado urina escura, icterícia e uma perda de peso de 60 quilos. O paciente foi internado para o trabalho e tratamento de sua condição não diagnosticada. A sua equipa internada estava preocupada com a possível doença de Wilson e encaminhou o paciente para a clínica de oftalmologia para avaliação dos anéis Kayser-Fleischer. Ele não teve alterações na visão ou outras queixas oculares.note-se que uma biopsia ao fígado aos 14 anos, realizada depois que ele foi encontrado para ter exames de rotina anormais antes de uma amassilectomia, mostrou fígado gordo que se pensava estar relacionado com o seu peso. Ele não teve seguimento adicional.história Ocular anterior: nenhuma história clínica: o doente fez uma biópsia hepática aos 14 anos de idade com um diagnóstico de fígado gordo. Ele também fez uma amígdala aos 14 anos. história familiar: não há história familiar de doença hepática. A mãe do paciente era alcoólica.História Social: O paciente está preso por roubo. Ele relata o uso de álcool pesado (mais de um pacote de seis cervejas por dia por mais de oito anos). Ele fuma um maço de cigarros por dia e relata o uso prévio de maconha, LSD e cocaína no passado. revisão dos sistemas: o paciente relatou uma perda involuntária de peso de 60 libras. Ele não tinha alterações de humor, alterações comportamentais ou distúrbios de movimento.exame Ocular: melhor acuidade visual corrigida: 20/20 e 20/20 OS. pupilas: 6 mm na escuridão, 3 mm na luz, sem RAPD OU: 12 mmHg OD e 14 mmHg OS
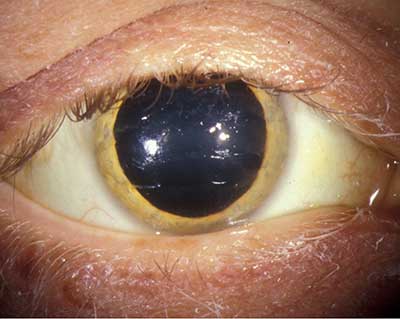
| Figure 1a: Golden Brown Kayser-Fleischer ring | Figure 1b: Maior ampliação da foto de marrom dourado anel no nível de Descemet da membrana |
 |
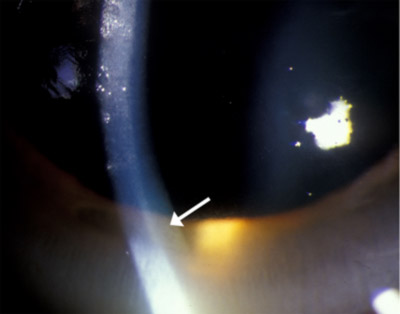 |
| Figura 1c: A gonioscopia de ângulo, mostrando dourar depósito de Descemet da membrana | |
 |
|
Curso
O paciente foi admitido para o UIHC para um diagnóstico do trabalho, bem como o tratamento para a insuficiência hepática grave. O trabalho subsequente revelou agravamento dos testes da função hepática. Tinha níveis séricos normais de ceruloplasmina e de cobre, mas tinha um nível elevado de cobre na urina de 189 ng/ml (o normal é 12-80) e uma excreção elevada de cobre na urina de 1522 mcg / dia (o normal é 3-35) após o teste de excreção de penicilamina. Sua biópsia hepática revelou uma quantificação hepática de cobre de 763 mcg de cobre por grama de peso seco (normal é 10-35 mcg/gm). devido à presença de anéis Kayser-Fleischer, elevada excreção de cobre na urina após teste de excreção de penicilamina e biópsia hepática de diagnóstico, o doente foi diagnosticado com a doença de Wilson. Foi então tratado com 250 mg de penicilamina 4 vezes por dia e 25 mg de piridoxina por dia, o que melhorou a sua depuração de cobre na urina. No entanto, ele desenvolveu encefalopatia significativa, um agravamento progressivo da coagulopatia com hemólise e anemia de doença hepática, e ascite significativa. Sua doença hepática continuou a progredir ao longo de seu curso de duas semanas no hospital, e ele demonstrou um curso sub-fulminante. Seus hepatologistas acharam que a doença poderia progredir para a insuficiência hepática e provável morte. O transplante de fígado foi incapaz de ser realizado em UIHC devido a sua história de uso de álcool e sua rede de suporte pobre. Ele foi então transferido para um hospital mais próximo de sua família.
discussão: a doença de Wilson
a doença de Wilson, descrita pela primeira vez em 1912 por Kinnear Wilson, é uma doença autossómica recessiva rara de excreção biliar de cobre e deposição de cobre em todo o corpo, mais notavelmente no fígado, cérebro, rins e córnea. A doença é causada por uma mutação genética no gene ATP7B no cromossomo 13 que codifica uma membrana de cobre transportando ATPase encontrada principalmente no fígado (Tanzi, 1993). Pacientes com doença de Wilson podem apresentar doença crônica do fígado, insuficiência hepática fulminante, insuficiência renal aguda, anemia hemolítica, ou neuropsiquiátricos de doenças como distúrbios do movimento, tremores, incoordenação, e alterações comportamentais (Ver Tabela 1). A doença pode apresentar-se em qualquer idade, mas é geralmente observada entre a primeira e a quarta décadas de vida (Schoen, 1990). A prevalência mundial foi relatada como sendo 1 em 30.000 (Scheinberg, 1984). Se não for tratada, esta doença é letal. Tratamentos comuns com a penicilamina, trientina e / ou terapia de zinco (tratamentos quelantes de cobre), bem como dietas de cobre baixas são esforços de vida longa, com o transplante de fígado sendo Salvador de vida em casos muito avançados (Mak, 2008).
| Hepática | Neurológicas | Psiquiátrica | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Icterícia Hepatite Aguda Cirrose Doença Hepática Crônica Insuficiência Hepática fulminante Insuficiência Renal Aguda Anemia Hemolítica |
Tremor a Distonia Bradicinesia Incoordenação Insônia a Espasticidade Coreia Babando Apreensões Enxaqueca |
Esquizofrenia Depressão alterações da personalidade alterações da personalidade alterações da personalidade alterações da personalidade alterações da personalidade alterações da personalidade em doentes com idade inferior a 40 anos com doença hepática inexplicável, doença neurológica comportamental e/ou psiquiátrica no contexto de doença hepática, ou historial familiar da doença de Wilson. O diagnóstico da doença de Wilson é muitas vezes baseado nos critérios de Sternlieb, onde um paciente deve ter pelo menos dois dos seguintes achados: a presença de anéis Kayser-Fleischer, sintomas neurológicos típicos e/ou baixos níveis de ceruloplasmina (<0,20 g/L) (Sternlieb, 1990). Infelizmente, estes critérios só são frequentemente cumpridos quando um doente apresenta uma doença avançada e geralmente apresenta manifestações neurológicas e / ou psiquiátricas. Doentes com doença precoce, doença hepática apenas, ou doença assintomática são muito difíceis de diagnosticar. não existem testes laboratoriais que identifiquem fiavelmente a doença de Wilson, mas uma combinação de testes incluindo ceruloplasmina sérica, cobre isento de soro e excreção urinária de cobre de 24 horas, são utilizados em conjunto para identificar anomalias no metabolismo do cobre. a ceruloplasmina é uma proteína produzida por hepatócitos que se liga ao cobre e a liberta nos tecidos periféricos. Quando o cobre não está disponível, devido ao transporte transmembranar deficiente, para ser incorporado na molécula de apoceruloplasmina como na doença de Wilson, a apoceruloplasmina libertada é rapidamente metabolizada, fazendo com que os níveis circulantes de ceruloplasmina sejam baixos. O nível normal de ceruloplasmina no sangue é de 0,20 a 0,40 g/L, no entanto, este valor não é aplicável a pacientes pediátricos, pacientes grávidas ou pacientes em estrogênio. Outras doenças podem causar baixos níveis, incluindo desnutrição, síndrome nefrótico, aceruloplasminemia hereditária, ou distúrbios inflamatórios (como é um reagente de fase aguda). Além disso, os níveis de ceruloplasmina podem ser normais em pacientes com doença de Wilson, citados como em qualquer lugar de 35% -45% em pacientes com apresentação hepática e 60% de pacientes com insuficiência hepática fulminante (Steindl, 1997). Portanto, em pacientes com doença hepática, um nível normal de ceruloplasmina não pode excluir a doença de Wilson, nem um nível baixo é suficiente para fazer um diagnóstico da doença de Wilson.
24 horas urinária de cobre excreção é sempre elevada em pacientes com doença de Wilson (>100 µg/dia ou 1.0 mol / dia), mas os resultados precisos são difíceis de obter, uma vez que a conformidade pode ser baixa, pode ocorrer uma recolha incompleta e pode ocorrer contaminação por cobre exógeno, como quando o recipiente de recolha de urina é lavado com água da torneira. A medição da excreção urinária de cobre de 24 horas antes e depois da administração de D-penacilamina pode ajudar a diferenciar a excreção de doente com Wilson de doentes com outras perturbações hepáticas, uma vez que os doentes com Wilsons terão uma excreção superior a 25 mol/dia. Este teste demonstrou ter uma sensibilidade de 76-88% e especificidade de 93-98% (Martins da Costa, 1992 e Muller, 2007). Este teste não foi, no entanto, confiável para diagnosticar pacientes assintomáticos com a doença de Wilson (sensibilidade de 46%) e é bastante complicado para os pacientes a realizar (Muller, 2007).mais recentemente, foram notificados casos de Utilização da fosfatase alcalina para a bilirrubina total e/ou da AST para ALT para ajudar no diagnóstico da doença de Wilson Em caso de insuficiência hepática aguda. A publication by Korman et al. reports that an alkaline phosphatase to total bilirrubin ratio of less than 4 yielded 94% sensitivity and 96% specificity with a likely ratio of 23 for diagnosing fulminant Wilson’s disease. Eles também relatam que uma razão AST:ALT superior a 2,2 foi 94% sensível e 86% específica com uma taxa de probabilidade de 7 para diagnosticar a doença de fulminante Wilson, e quando os testes foram combinados, a sensibilidade diagnóstica e especificidade foi 100% (Korman, 2008).o teste padrão ouro para diagnosticar a doença de Wilson está medindo o conteúdo de cobre hepático em uma biópsia hepática. De acordo com Ferenci et al. um teor de cobre de >250 µg/g de peso seco (normal, até 50 µg / g de peso seco) é 83% sensível e 98,6% específico para o diagnóstico da doença de Wilson, mas realizar biópsias hepáticas é um procedimento invasivo e arriscado em pacientes com insuficiência hepática grave que têm coagulopatia (Ferenci, 2005).o teste genético do gene ATP7B pode ser realizado com elevada sensibilidade se a mutação for conhecida de um proband ou se o doente tiver uma das mutações genéticas comuns para a doença de Wilson. No entanto, o teste genético não é rotineiramente utilizado como uma ferramenta de diagnóstico, como o teste é complicado de executar devido ao gene de longa duração (21 éxons), suas inúmeras mutações (mais de 70 diferentes mutações), e o fato de que a maioria dos pacientes tem duas mutações diferentes (composto heterozigotos) (Mak, 2008). devido à dificuldade em diagnosticar a doença de Wilson, um sistema de pontuação foi criado e promovido pelo 8º Encontro Internacional sobre a doença de Wilson, que é baseado em sete critérios, incluindo a presença de anéis Kayser-Flesicher; sintomas neurológicos típicos; diminuição da concentração sérica de ceruloplasmina; anemia hemolítica negativa de Coombs; elevada excreção urinária de cobre; elevado valor hepático de cobre na ausência de colestase; e resultados mutacionais. Como todos os outros testes laboratoriais, este sistema de pontuação tende a ser mais confiável em pacientes com doença avançada (Ferenci, 2003). o anel Kayser-Fleischer é a marca da doença de Wilson e sua detecção pode ser crítica para o diagnóstico. Há relatos de que foi a primeira manifestação detectável da doença de Wilson, que levou ao diagnóstico precoce e tratamento da doença (Liu, 2002). a presença de anéis Kayser-Fleischer em combinação com baixa ceruloplasmina sérica é considerada diagnóstico da doença de Wilson com base nos critérios de Sternlieb (Martins da Costa, 1992). Na córnea, o excesso de cobre circulante é depositado na membrana de Descemet e é geralmente visto como um anel castanho dourado localizado na córnea periférica, começando na linha de Schwalbe e estendendo-se menos de 5 mm sobre a córnea (ver vídeo). O anel também pode aparecer como amarelo esverdeado, vermelho rubi, verde brilhante ou azul ultramarino. É quase sempre bilateral e aparece superiorly primeiro, então inferiorly, e depois torna-se circunferencial (Kim, 1979). Nos estágios iniciais da doença, a Gonioscopia é muitas vezes necessária para detectar este achado sutil, mas na doença avançada ela pode ser vista a olho nu. Vídeo 1: Marrom Dourado Kayser anel de Fleischer link alternativo para dourar Kayser anel de Fleischer Estes anéis foram relatados para ser visto em cerca de 85% a 100% dos pacientes com neurológicos e/ou psiquiátricos manifestações da doença de Wilson, mas apenas 33%-86% dos pacientes com doença hepática e 0%-59% de pacientes assintomáticos (Mak, 2008). Anéis Kayser-Fleischer podem estar ausentes em até 50% dos pacientes com doença hepática Wilsoniana e em uma proporção ainda maior com doença hepática fulminante Wilsonian (Steindl, 1997). Há um número de condições que também têm sido ligados a anéis coloridos na córnea, incluindo outras doenças do fígado, como a cirrose biliar primária, hepatite neonatal, e cirrose cirrose, ou elevadas de cobre por outros motivos, tais como mieloma múltiplo, carcinoma pulmonar, gamopatias monoclonais benignas, leucemia linfocítica crônica, ou mesmo o uso de contraceptivos orais. Após o início do tratamento, o anel Kayser-Fleischer desaparece em 85-90% dos casos (Lossner, 1986).a doença de Wilson na UIHC na Universidade de Iowa, a clínica de Oftalmologia de plantão e o serviço de internamento frequentemente recebem consultas para avaliar os anéis de Kayser-Fleischer em pacientes com doença hepática inexplicável, mas poucos oftalmologistas já viram um verdadeiro anel de Kayser Fleischer. Em uma pesquisa realizada de todos os participantes, companheiros e residentes na UIHC, apenas 11 médicos relataram ter visto um anel Kayser-Fleischer (41%), com um total de casos relatados sendo 24 ao longo de toda a carreira de todos os oftalmologistas pesquisados. Muitas vezes, os anéis Kayser-Fleischer de um paciente foram vistos por vários oftalmologistas no departamento de modo que o número total de pacientes diagnosticados é menor do que o número total de casos relatados vistos. Dos que tinham visto um anel Kayser-Fleischer, apenas seis oftalmologistas tinham visto mais de um caso. Para 64% dos médicos, a identificação de um anel Kayser-Fleischer foi útil no diagnóstico da doença de Wilson em pelo menos um caso. uma pesquisa de seis hepatologistas na Universidade de Iowa, um centro de cuidados terciários, revelou apenas quatro novos diagnósticos da doença de Wilson em um período médio de prática clínica de 13 anos por médico. De acordo com a Pesquisa, nenhum dos casos relatados tinha um anel Kayser-Fleischer. O serviço de Neurologia também foi pesquisado sobre a sua experiência no diagnóstico da doença de Wilson. Um neurologista, que tem praticado por 37 anos, tem visto apenas um paciente com a doença de Wilson, e o paciente tinha um anel Kayser-Fleischer. Um outro neurologista que participou da pesquisa praticou por sete anos e não viu pacientes com a doença de Wilson. custo de diagnosticar a doença de Wilson em UIHC:com o custo crescente dos cuidados de saúde em mente, uma investigação sobre o custo médio de diagnosticar um paciente com a doença de Wilson em UIHC foi realizada. Consulta Especializada, bem como investigação laboratorial é necessária para fazer o diagnóstico na maioria dos casos. Apenas para a avaliação laboratorial básica, o custo é de US $190 (AST, ALT, fosfatase alcalina, bilirrubina Total e ceruloplasmina sérica), não incluindo os outros testes laboratoriais que normalmente seriam realizados em um paciente com qualquer doença hepática, incluindo hemoglobina, plaquetas, painel metabólico básico, e níveis de albumina (Ver quadro 3a). Em casos de dificuldade diagnóstica (apresentação inicial ou doença hepática), o diagnóstico pode custar até us $605 para o laboratório de avaliação (incluindo testes de função hepática, ceruloplasmina, e 24 horas urinária de cobre excreção), além de ambulatório de consultas taxas para oftalmologia ($300), hepatologia ($935), e/ou neurologia (us$775), para um custo total de us $2,615 (Ver Tabelas 3a, 3b e 3c). O custo total para a realização da biópsia hepática padrão ouro é de US $3,105, que inclui a taxa de médico, taxa de hospital, taxa de patologia, bem como o valor de enviar laboratório para o conteúdo de cobre hepático. No total, se todos os testes laboratoriais, serviços de consulta e biópsia hepática fossem utilizados, o custo total seria $ 5,720 (Ver Tabela 3c
|