Una Revisión Basada en Sistemas del Papel del Oftalmólogo en el Diagnóstico de la Enfermedad de Wilson
Emily S. Birkholz, MD, Thomas A. Oetting, MS, MD
28 de julio de 2009
Queja Principal: Remisión para evaluación de Anillos Kayser-Fleischer
Antecedentes de Enfermedad Presente: El paciente era un varón de 25 años que había estado experimentando sensibilidad subcostal del lado derecho y fatiga creciente durante los últimos meses. También había notado orina oscura, ictericia y pérdida de peso de 60 libras. El paciente fue ingresado para un examen y tratamiento de su condición no diagnosticada. Su equipo de pacientes hospitalizados estaba preocupado por la posible enfermedad de Wilson y remitió al paciente a la clínica de oftalmología para la evaluación de los anillos de Kayser-Fleischer. No tenía cambios en la visión u otras quejas oculares.
Cabe destacar que una biopsia de hígado a los 14 años, realizada después de que se comprobara que tenía pruebas de laboratorio de rutina anormales antes de una amigdalectomía, mostró hígado graso que se pensaba que estaba relacionado con su peso. No tuvo seguimiento adicional.
Antecedentes oculares anteriores: Ninguno
Antecedentes médicos: El paciente se sometió a una biopsia hepática a los 14 años con diagnóstico de hígado graso. También se sometió a una amigdalectomía a los 14 años.
Medicamentos: Ninguno
Antecedentes familiares: No hay antecedentes familiares de enfermedad hepática. La madre del paciente era alcohólica.
Historia social: El paciente está actualmente encarcelado por robo. Informa de un consumo excesivo de alcohol (más de un paquete de seis cervezas al día durante más de ocho años). Fuma un paquete de cigarrillos al día y reporta el uso previo de marihuana, LSD y cocaína en el pasado.
Revisión de sistemas: El paciente reportó una pérdida de peso involuntaria de 60 libras. No tenía cambios de humor, cambios de comportamiento o trastornos del movimiento.
Examen ocular:
- Agudeza visual mejor corregida: 20/20 OD y 20/20 SG.
- Pupilas: 6 mm en la oscuridad, 3 mm en la luz, sin RAPD OU
- PIO: DO de 12 mmHg y SG de 14 mmHg
- MOE: Unidad organizativa completa
- CVF: Unidad organizativa completa
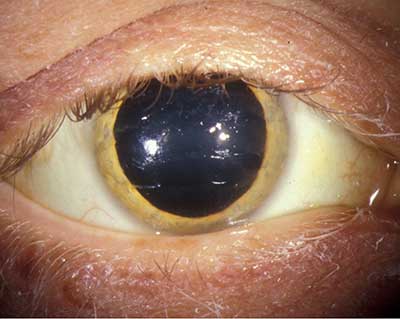
- Segmento anterior ver Figura 1. Ictericia escleral OU, banda marrón dorada de 1-2 mm vista en la OU limbus.
- DFE: discos normales, mácula normal, periferia y vasos OU.
| Figura 1a: Anillo dorado de Kayser-Fleischer marrón | Figura 1b: Foto de mayor aumento del anillo marrón dorado a nivel de la membrana de Descemet |
 |
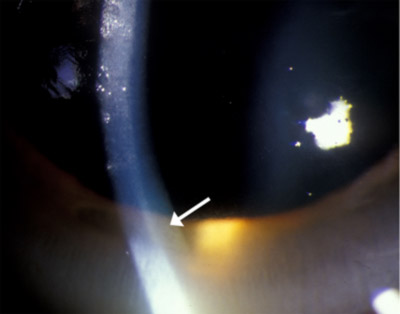 |
| Figura 1c: Gonioscopia de ángulo, que muestra un depósito marrón dorado en la membrana de Descemet | |
 |
|
Curso
El paciente fue admitido en el UIHC para un estudio de diagnóstico, así como para el tratamiento de la insuficiencia hepática grave. El estudio posterior reveló un empeoramiento de las pruebas de la función hepática. Tenía niveles séricos normales de ceruloplasmina y cobre, pero tenía un cobre urinario elevado de 189 ng/ml (normal es de 12-80), y una excreción de cobre urinario elevada de 1522 mcg / día (normal es de 3-35) después de la prueba de excreción de penicilamina. Su biopsia hepática reveló una cuantificación de cobre hepático de 763 mcg de cobre por gramo de peso seco (lo normal es de 10-35 mcg/gm).
Debido a la presencia de anillos de Kayser-Fleischer, excreción elevada de cobre en orina después de la prueba de excreción de penicilamina y biopsia hepática diagnóstica, el paciente fue diagnosticado con enfermedad de Wilson. Fue entonces tratados con penicilamina 250 mg 4 veces al día y piridoxina 25 mg PO diaria que mejoró su orina de cobre de separación. Sin embargo, desarrolló una encefalopatía significativa, una coagulopatía que empeoraba progresivamente con hemólisis y anemia de enfermedad hepática, y ascitis significativa. Su enfermedad hepática continuó progresando durante su curso de dos semanas en el hospital, y demostró un curso sub-fulminante. Sus hepatólogos consideraron que la enfermedad probablemente progresaría a insuficiencia hepática y probablemente la muerte. El trasplante de hígado no se pudo realizar en la UIHC debido a su historial de consumo de alcohol y su pobre red de apoyo. He was then transferred to a hospital closer to his family.
Discusión: Enfermedad de Wilson
La enfermedad de Wilson, descrita por primera vez en 1912 por Kinnear Wilson, es una enfermedad autosómica recesiva rara de excreción biliar de cobre y deposición de cobre en todo el cuerpo, especialmente en el hígado, el cerebro, los riñones y la córnea. La enfermedad es causada por una mutación genética en el gen ATP7B en el cromosoma 13 que codifica para una ATPasa transportadora de cobre unida a membrana que se encuentra principalmente en el hígado (Tanzi, 1993). Los pacientes con enfermedad de Wilson pueden presentar enfermedad hepática crónica, insuficiencia hepática fulminante, insuficiencia renal aguda, anemia hemolítica o enfermedad neuropsiquiátrica como trastornos del movimiento, temblores, falta de coordinación y cambios de comportamiento (Ver Tabla 1). La enfermedad puede presentarse a cualquier edad, pero generalmente se observa entre las décadas 1 y 4 de la vida (Schoen, 1990). Se ha informado que la prevalencia mundial es de 1 de cada 30.000 (Scheinberg, 1984). Si no se trata, esta enfermedad es letal. Los tratamientos comunes con penicilamina, trientina y / o terapia de zinc (tratamientos de quelación de cobre), así como las dietas bajas en cobre, son esfuerzos de por vida, con el trasplante de hígado salvando vidas en casos muy avanzados (Mak, 2008).
| Hepática | Neurológica | Psiquiátrica |
|---|---|---|
|
Ictericia Hepatitis Aguda Cirrosis Enfermedad Hepática Crónica Insuficiencia Hepática Fulminante Insuficiencia Renal aguda Anemia Hemolítica |
Temblor Distonía Bradicinesia Incoordinación Insomnio Espasticidad Corea Babeo Convulsiones Migraña |
Esquizofrenia Depresión Trastorno Maníaco Depresivo Delirios Trastornos del comportamiento Cambios de personalidad |
El diagnóstico de la enfermedad de Wilson
La enfermedad de Wilson generalmente se sospecha en pacientes jóvenes menores de cuarenta años que tienen enfermedad hepática inexplicable, enfermedad neurológica conductual inexplicable y/o enfermedad psiquiátrica en el entorno de enfermedad hepática o antecedentes familiares de enfermedad de Wilson. El diagnóstico de la enfermedad de Wilson a menudo se basa en los criterios de Sternlieb, donde un paciente debe tener al menos dos de los siguientes hallazgos: la presencia de anillos de Kayser-Fleischer, síntomas neurológicos típicos y/o niveles bajos de ceruloplasmina (<0,20 g/L) (Sternlieb, 1990). Desafortunadamente, estos criterios a menudo solo se cumplen cuando un paciente presenta una enfermedad avanzada y, por lo general, tiene manifestaciones neurológicas y/o psiquiátricas. Los pacientes con enfermedad temprana, enfermedad hepática solamente o enfermedad asintomática son muy difíciles de diagnosticar.
No existe ninguna prueba de laboratorio que identifique de manera confiable la enfermedad de Wilson, pero una combinación de pruebas que incluyen ceruloplasmina sérica, cobre libre de suero y excreción urinaria de cobre de 24 horas se utilizan juntas para identificar anomalías en el metabolismo del cobre.
La ceruloplasmina es una proteína elaborada por los hepatocitos que se une al cobre y lo transporta a los tejidos periféricos. Cuando el cobre no está disponible, debido a un transporte transmembrana deficiente, para ser incorporado a la molécula de apoceruloplasmina, como en la enfermedad de Wilson, la apoceruloplasmina liberada se metaboliza rápidamente, causando que los niveles circulantes de ceruloplasmina sean bajos. El nivel normal de ceruloplasmina en la sangre es de 0,20 a 0,40 g / L, sin embargo, este valor no es aplicable a pacientes pediátricos, pacientes embarazadas o pacientes con estrógeno. Otras enfermedades pueden causar niveles bajos, como desnutrición, síndrome nefrótico, aceruloplasminemia hereditaria o trastornos inflamatorios (ya que es un reactivo de fase aguda). Además, los niveles de ceruloplasmina pueden ser normales en pacientes con enfermedad de Wilson, citados entre el 35 y el 45% en pacientes con presentación hepática y el 60% en pacientes con insuficiencia hepática fulminante (Steindl, 1997). Por lo tanto, en pacientes con enfermedad hepática, un nivel normal de ceruloplasmina no puede excluir la enfermedad de Wilson, ni un nivel bajo es suficiente para hacer un diagnóstico de la enfermedad de Wilson.
- Anillos Kayser-Fleischer
- Niveles bajos de Ceruloplasmina sérica (<0,20 g/l, normal es de 0,20 a 0,40 g/l)
- Excreción Urinaria de cobre de 24 horas (>100 µg/día o 1.0 mol/día)
- Excreción Urinaria de Cobre durante 24 horas después de D-penacilamina (>25 mol/día)
- Contenido hepático de cobre en biopsia hepática (>250 µg/g de peso seco, normal es< 50 µg/g de peso seco)
- Mutación genética en el gen ATP7B
La excreción urinaria de cobre de 24 horas siempre es elevada en pacientes con enfermedad de Wilson (>100 µg/día o 1.0 mol / día), pero es difícil obtener resultados precisos, ya que el cumplimiento puede ser bajo, la recolección incompleta puede ocurrir y la contaminación del cobre exógeno puede ocurrir como cuando el recipiente de recolección de orina se enjuaga con agua del grifo. Medir la excreción urinaria de cobre de 24 horas antes y después de la administración de D-penacilamina puede ayudar a diferenciar a los pacientes con Wilson de los pacientes con otros trastornos hepáticos, ya que los pacientes con Wilson tendrán una excreción de más de 25 mol/día. Se ha demostrado que esta prueba tiene una sensibilidad del 76-88% % y una especificidad del 93-98% (Martins da Costa, 1992 y Muller, 2007). Sin embargo, esta prueba no fue confiable para diagnosticar pacientes asintomáticos con enfermedad de Wilson (sensibilidad del 46%) y es bastante engorrosa para que los pacientes la realicen (Muller, 2007).
Más recientemente, ha habido informes de la utilización de la relación fosfatasa alcalina a bilirrubina total y / o la relación AST a ALT para ayudar en el diagnóstico de la enfermedad de Wilson en el contexto de insuficiencia hepática aguda. Una publicación de Korman et al. se informa que una proporción de fosfatasa alcalina a bilirrubina total de menos de 4 produjo una sensibilidad de 94% y una especificidad de 96%, con una proporción de probabilidad de 23 para el diagnóstico de la enfermedad de Wilson fulminante. También informan que una relación AST: ALT de más de 2,2 fue 94% sensible y 86% específica, con una relación de probabilidad de 7 para el diagnóstico de la enfermedad de Wilson fulminante, y cuando se combinaron las pruebas, la sensibilidad diagnóstica y la especificidad fueron del 100% (Korman, 2008).
La prueba estándar de oro para diagnosticar la enfermedad de Wilson es medir el contenido de cobre hepático en una biopsia hepática. Según Ferenci et al. un contenido de cobre de >250 µg/g de peso seco (normal, hasta 50 µg/g de peso seco) es 83% sensible y 98,6% específico para diagnosticar la enfermedad de Wilson, pero realizar biopsias hepáticas es un procedimiento invasivo y arriesgado en pacientes con insuficiencia hepática grave que tienen coagulopatía (Ferenci, 2005).
Las pruebas genéticas del gen ATP7B se pueden realizar con alta sensibilidad si la mutación se conoce a partir de un probanda o si el paciente tiene una de las mutaciones genéticas comunes para la enfermedad de Wilson. Sin embargo, las pruebas genéticas no se utilizan rutinariamente como herramienta de diagnóstico, ya que las pruebas son engorrosas debido a la larga longitud del gen (21 exones), sus numerosas mutaciones (más de 70 mutaciones diferentes) y el hecho de que la mayoría de los pacientes tienen dos mutaciones diferentes (heterocigotos compuestos) (Mak, 2008).
Debido a la dificultad en el diagnóstico de la enfermedad de Wilson, se creó y promovió un sistema de puntuación en la 8a reunión Internacional sobre la enfermedad de Wilson que se basa en siete criterios, incluida la presencia de anillos Kayser-Flesicher; síntomas neurológicos típicos; disminución de la concentración sérica de ceruloplasmina, anemia hemolítica negativa de Coombs, excreción urinaria elevada de cobre, valor elevado de cobre hepático en ausencia de colestasis y hallazgos mutacionales. Al igual que todas las demás pruebas de laboratorio, este sistema de puntuación tiende a ser más fiable en pacientes con enfermedad avanzada (Ferenci, 2003).
Anillo de Kayser-Fleischer
El anillo de Kayser-Fleischer es el sello distintivo de la enfermedad de Wilson y su detección puede ser crítica para el diagnóstico. Hay informes en los que ha sido la primera manifestación detectable de la enfermedad de Wilson, que llevó al diagnóstico y tratamiento tempranos de la enfermedad (Liu, 2002).
La presencia de anillos de Kayser-Fleischer en combinación con ceruloplasmina sérica baja se considera un diagnóstico de la enfermedad de Wilson basado en los criterios de Sternlieb (Martins da Costa, 1992). En la córnea, el exceso de cobre circulante se deposita en la membrana de Descemet y generalmente se ve como un anillo marrón dorado ubicado en la córnea periférica, comenzando en la línea de Schwalbe y extendiéndose menos de 5 mm sobre la córnea (ver video). El anillo también puede aparecer como amarillo verdoso, rojo rubí, verde brillante o azul ultramarino. Casi siempre es bilateral y aparece primero por encima, luego por debajo, y luego se vuelve circunferencial (Kim, 1979). En las primeras etapas de la enfermedad, a menudo se necesita una gonioscopia para detectar este hallazgo sutil, pero en la enfermedad avanzada se puede ver a simple vista.
Video 1: Anillo de Kayser Fleischer marrón dorado
Enlace alternativo para el anillo de Kayser Fleischer marrón dorado
Se ha informado que estos anillos se observan en aproximadamente el 85-100% de los pacientes con manifestaciones neurológicas y / o psiquiátricas de la enfermedad de Wilson, pero solo en el 33-86% de los pacientes con enfermedad hepática y en el 0-59% de los pacientes asintomáticos (Mak, 2008). Los anillos de Kayser-Fleischer pueden estar ausentes en hasta el 50% de los pacientes con enfermedad hepática wilsoniana y en una proporción aún mayor con enfermedad hepática wilsoniana fulminante (Steindl, 1997). Hay una serie de afecciones que también se han relacionado con anillos de colores en la córnea, incluidas otras enfermedades hepáticas como cirrosis biliar primaria, hepatitis neonatal y cirrosis criptogénica, o cobre elevado por otras razones, como el mieloma múltiple, el carcinoma pulmonar, las gammopatías monoclonales benignas, la leucemia linfocítica crónica o incluso el uso de anticonceptivos orales. Tras el inicio del tratamiento, el anillo Kayser-Fleischer desaparece en el 85-90% de los casos (Lossner, 1986).
Enfermedad de Wilson en UIHC
En la Universidad de Iowa, la clínica de oftalmología de guardia y el servicio de hospitalización reciben consultas frecuentes para evaluar anillos Kayser-Fleischer en pacientes con enfermedad hepática inexplicable, pero pocos oftalmólogos han visto nunca un anillo Kayser Fleischer verdadero. En una encuesta realizada a todos los asistentes, becarios y residentes de UIHC, solo 11 médicos informaron haber visto un anillo Kayser-Fleischer (41%), con un total de casos reportados de 24 a lo largo de toda la carrera de todos los oftalmólogos encuestados. A menudo, los anillos de Kayser-Fleischer de un paciente fueron vistos por múltiples oftalmólogos en el departamento, por lo que el número total de pacientes diagnosticados es menor que el número total de casos notificados vistos. De los que habían visto un anillo Kayser-Fleischer, solo seis oftalmólogos habían visto más de un caso. Para el 64% de los médicos, la identificación de un anillo de Kayser-Fleischer fue útil en el diagnóstico de la enfermedad de Wilson en al menos un caso.
Una encuesta de seis hepatólogos de la Universidad de Iowa, un centro de atención terciaria, reveló solo cuatro nuevos diagnósticos de la enfermedad de Wilson en un período medio de práctica clínica de 13 años por médico. Según la encuesta, ninguno de los casos reportados tenía un anillo Kayser-Fleischer.
El servicio de neurología también fue encuestado sobre su experiencia en el diagnóstico de la enfermedad de Wilson. Un neurólogo, que ha ejercido durante 37 años, solo ha visto a un paciente con enfermedad de Wilson, y el paciente tenía un anillo de Kayser-Fleischer. Otro neurólogo que participó en la encuesta ha ejercido durante siete años y no ha visto a ningún paciente con la enfermedad de Wilson.
Costo del diagnóstico de la enfermedad de Wilson en el UIHC:
Teniendo en cuenta el costo cada vez mayor de la atención médica, se realizó una investigación sobre el costo promedio del diagnóstico de un paciente con enfermedad de Wilson en el UIHC. En la mayoría de los casos, se requiere una consulta especializada, así como una investigación de laboratorio para hacer el diagnóstico. Solo para la evaluación básica de laboratorio, el costo es de 1 190 (AST, ALT, fosfatasa alcalina, Bilirrubina Total y ceruloplasmina sérica), sin incluir las otras pruebas de laboratorio que normalmente se realizarían en un paciente con cualquier enfermedad hepática, como hemoglobina, plaquetas, panel metabólico básico y niveles de albúmina (Ver Tabla 3a). En casos de dificultad diagnóstica (presentación temprana o enfermedad hepática solamente), el diagnóstico puede costar hasta 6 605 para la evaluación de laboratorio (incluidas pruebas de función hepática, ceruloplasmina y excreción urinaria de cobre de 24 horas), además de los honorarios de consulta ambulatoria para oftalmología (3 300), hepatología (9 935) y/o neurología (7 775), por un costo total de 2 2,615 (Ver Tablas 3a, 3b y 3c). El costo total para realizar la biopsia hepática estándar de oro es de 3 3,105, que incluye la tarifa médica, la tarifa hospitalaria, la tarifa de patología, así como el valor de laboratorio enviado para el contenido de cobre hepático. En total, si se utilizaran todas las pruebas de laboratorio, los servicios de consulta y la biopsia hepática, el costo total sería de 5 5,720 (Consulte la Tabla 3c
| Lab Test | Cost |
|---|---|
| AST | $29 |
| ALT | $34 |
| ALP | $32 |
| Total Bilirubin | $29 |
| Serum Ceruloplasmin | $66 |
| 24-h urine copper excretion | $70 |
| Hepatic Copper Content (from liver biopsy send out lab) |
$275 |
| Consult/Procedure | Cost |
|---|---|
| Ophthalmology outpatient consult (low-moderate complexity) | $300 |
| Ophthalmology inpatient consult (low-moderate complexity) | $280 |
| Neurology Outpatient consult (moderate-high complexity) | $775 |
| Neurology Inpatient Consultation (moderate-high complexity) | $650 |
| Hepatology Outpatient Consultation (moderate-high complexity) | $935 |
| Hepatology Inpatient Consultation (moderate-high complexity) | $860 |
| Liver Biopsy: Total (outpatient costs) | $2,830 |
| Physician charge | $1,226 |
| Hospital charge | $926 |
| Pathology professional fee | $312 |
| Pathology technical fee | $366 |
| services | cost |
|---|---|
|
Diagnosis without ophthalmology consultation |
$2,315 |
|
Diagnosis with ophthalmology consultation |
$2,615 |
|
Total cost of liver biopsy |
$3,105 |
|
Diagnosis with all common tests, consults, and biopsia hepática |
$5,720 |
Resumen
Aunque es extremadamente raro en UIHC con solo cuatro o cinco nuevos diagnósticos de enfermedad de Wilson en un período de tiempo promedio de 13 años basado en una encuesta de hepatólogos, una evaluación oftalmológica para buscar anillos Kayser-Fleischer sigue siendo una herramienta de diagnóstico muy esencial y es una forma no invasiva y asequible de ayudar en el diagnóstico de una enfermedad potencialmente mortal. Sin embargo, pocos oftalmólogos tienen experiencia en identificar un anillo Kayser-Fleischer, ya que solo el 41% de los oftalmólogos encuestados en la Universidad de Iowa han visto uno.
Cuando un paciente presenta una enfermedad avanzada o manifestaciones neurológicas y / o psiquiátricas de la enfermedad de Wilson, un anillo de Kayser-Fleischer está presente en casi todos los casos y puede ayudar de forma no invasiva a solidificar el diagnóstico. Cuando un paciente se presenta con enfermedad menos avanzada o enfermedad hepática solamente, el diagnóstico es mucho más difícil y a menudo se necesita la evaluación crítica de todas las pruebas disponibles para confirmar el diagnóstico. Debido a que muchas pruebas de laboratorio no son concluyentes en pacientes con enfermedad menos avanzada, y debido a que la biopsia hepática estándar de oro es un procedimiento invasivo y de alto costo, la evaluación no invasiva de un anillo de Kayser-Fleischer sigue siendo una parte esencial del trabajo de diagnóstico para la enfermedad de Wilson. En estos casos, identificar un anillo de Kayser-Fleischer a menudo es mucho más difícil, ya que el anillo puede ser muy débil e identificable solo en la gonioscopia. Por lo tanto, una evaluación en la clínica utilizando una lámpara de hendidura y una lente de gonioscopia es esencial en este grupo de pacientes.
- Aldave AJ, King JA, Kim BT, Hopp L. Depósito de cobre corneal asociado con leucemia linfocítica crónica. Am J Ophthalmol 2006; 142: 174-6.
- Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnóstico y clasificación fenotípica de la enfermedad de Wilson. Liver Int 2003; 23: 139-142.
- Ferenci P, Steindl-Munda P, VogelW, JessnerW, Gschwantler M, Stauber R, Datz C,Hackl F, Wrba F, Bauer P, Lorenz O. Valor diagnóstico de la determinación cuantitativa de cobre hepático en pacientes con enfermedad de Wilson. Clin Gastroenterol Hepatol 2005; 3: 811-818.
- Fleming CR, Dickson ER, Wahner HW, Hollenhorst RW, McCall JT. Anillos corneales pigmentados en enfermedad hepática no wilsoniana. Ann Intern Med 1977;86:285 – 8.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Anillos similares a Kayser-Fleischer en pacientes sin enfermedad de Wilson. Gastroenterología 1977;72:1331 – 5.
- Garmizo G, Frauens BJ. Depósito corneal de cobre secundario a anticonceptivos orales. Optom Vis Sci. Septiembre de 2008; 85 (9): E802-7.
- Hawkins AS, Stein RM, Gaines BI, Deutsch TA. Depósito ocular de cobre asociado con mieloma múltiple. Am J Ophthalmol 2001; 131: 257-9.
- Kim HB, Kim JC, Byan YJ. El anillo de Kayser Fleischer en la enfermedad de Wilson. J Oftal coreano Soc 1979;20:129-31.
- Korman JD, Volenber I, Balko J, et al. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests (en inglés). Hepatología. 2008;48(4): 1167-74.Liu M, Cohen EJ, Brewer GJ, Laibson PR. Anillo de Kayser-Fleischer como signo de presentación de la enfermedad de Wilson. Am J Ophthalmol. Junio de 2002;133 (6): 832-4.
- Lossner A, Lossner J, Bachmann H, Zotter J. El anillo de Kayser Fleischer durante el tratamiento a largo plazo en la enfermedad de Wilson (degeneración hepatolenticular): Un estudio de seguimiento. Graefes Arch Clin Exp Ophthalmol. 1986;224:152-5.
- Mak CM, Lam CW. Diagnosis of Wilson disease: a comprehensive review (en inglés). Revisiones Críticas en Ciencias de Laboratorio Clínico. 2008;45(3):263-290.
- Martin NF, Kincaid MC, Stark WJ, Petty BG, Surer JL, Hirst LW, Green WR. Depósito ocular de cobre asociado con carcinoma pulmonar, gammapatía monoclonal IgG e hipercupremia. Una correlación clinicopatológica. Ophthalmology 1983; 90: 110-16.
- Martins da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, Mieli-Vergani G. Valor de la excreción urinaria de cobre después del desafío de penicilamina en el diagnóstico de la enfermedad de Wilson. Hepatology 1992; 15: 609-615.
- Muller T, Koppikar S, Taylor RM, Carragher F, Schlenck B, Heinz-Erian P, Kronenberg F, Ferenci P, Tanner S, Siebert U, Staudinger R, Mieli-Vergani G, Dhawan A. Reevaluación de la prueba de desafío de penicilamina en el diagnóstico de la enfermedad de Wilson en niños. J Hepatol 2007; 47: 270 – 276.Probst LE, Hoffman E, Cherian MG, Yang J, Feagan B, Adams P, Nichols B. Depósito ocular de cobre asociado con gammapatía monoclonal benigna e hipercupremia. Córnea 1996; 15: 94-8.
- Enfermedad de Scheinberg I, Sternlieb I. Wilson. Major Probl Intern Med 1984; 23: 1-24.
- Schoen RE, Sternlieb I. Aspectos clínicos de la enfermedad de Wilson. Am J Gastroenterol -1990; 85: 1453-7.
- Steindl P, Ferenci P, Dienes HP. Enfermedad de Wilson en pacientes con enfermedad hepática: un desafío diagnóstico. Gastroenterology 1997; 113: 212 a 218.
- Sternlieb I. Perspectivas sobre la enfermedad de Wilson. Hepatología 1990; 12: 1234 – 1239.
- Tanzi RE, Petrukhin K, Chernov I, et al. El gen de la enfermedad de Wilson es una ATPasa transportadora de cobre con homología al gen de la enfermedad de Menkes. Nat Genet 1993; 5: 344-50.
Agradecimiento especial al Dr. Michael Voigt, Dr. Bruce Luxon, la Dra. Stephanie Dee, el Dr. Kyle Brown, el Dr. Douglas LaBrecque y el Dr. Warren Schmidt , todos forman parte de la división de Hepatología del departamento de Gastroenterología de los Hospitales y Clínicas de la Universidad de Iowa (UIHC). Gracias también al Dr. Robert Rodnitzky y al Dr. Pedro González en el departamento de Neurología de UIHC.
Formato de Cita sugerido: Birkholz ES, Oetting TA. Anillo de Kayser Fleischer: Una Revisión Basada en Sistemas del Papel del Oftalmólogo en el Diagnóstico de la Enfermedad de Wilson. EyeRounds.org. 28 de julio de 2009; Disponible en: http://www.EyeRounds.org/cases/97-kayser-fleischer-ring-Wilsons-Disease.htm.