o revizuire bazată pe sisteme a rolului oftalmologului în diagnosticul bolii Wilson
Emily S. Birkholz, MD, Thomas A. Oetting, MS, MD
28 iulie 2009
plângere șef: sesizare pentru evaluarea inelelor Kayser-Fleischer
istoricul bolii prezente: Pacientul era un bărbat în vârstă de 25 de ani, care se confrunta cu o sensibilitate subcostală pe partea dreaptă și o oboseală crescândă în ultimele câteva luni. De asemenea, a observat urină închisă la culoare, icter și o pierdere în greutate de 60 de kilograme. Pacientul a fost internat pentru muncă și tratamentul stării sale nediagnosticate. Echipa sa de internare a fost preocupată de posibila boală Wilson și a trimis pacientul la Clinica de oftalmologie pentru evaluarea inelelor Kayser-Fleischer. Nu a avut modificări ale vederii sau alte plângeri oculare.
de remarcat, o biopsie hepatică la vârsta de 14 ani, efectuată după ce s-a constatat că are laboratoare anormale de rutină înainte de o amigdalectomie, a arătat ficat gras despre care se credea că este legat de greutatea sa. Nu a avut nicio urmărire suplimentară.
antecedente oculare anterioare: niciunul
istoric Medical: pacientul a făcut o biopsie hepatică la vârsta de 14 ani cu un diagnostic de ficat gras. De asemenea, a avut o amigdalectomie la vârsta de 14 ani.
medicamente: niciunul
istoric familial: nu există antecedente familiale de boli hepatice. Mama pacientului era alcoolică.
istoria socială: Pacientul este în prezent încarcerat pentru furt. El raportează consumul intens de alcool (mai mult de un pachet de șase bere pe zi timp de peste opt ani). Fumează un pachet de țigări pe zi și raportează consumul anterior de marijuana, LSD și cocaină în trecut.
revizuirea sistemelor: pacientul a raportat o pierdere neintenționată în greutate de 60 de kilograme. Nu avea schimbări de dispoziție, schimbări de comportament sau tulburări de mișcare.
examen Ocular:
- cea mai bună acuitate vizuală corectată: 20/20 OD și 20/20 OS.
- elevi: 6 mm în întuneric, 3 mm în lumină, fără RAPD OU
- IOP: 12 mmHg OD și 14 mmHg OS
- MOA: full OU
- CVF: full ou
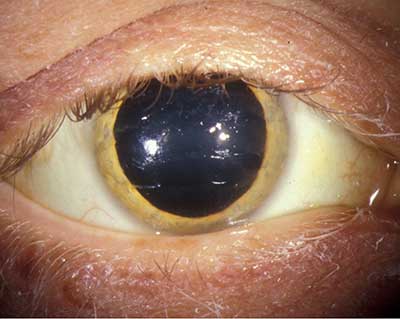
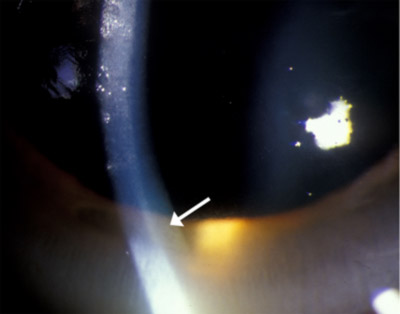
- segment Anterior a se vedea Figura 1. Scleral icterus OU, bandă maro aurie de 1-2 mm văzută la limbus OU.
- DFE: discuri normale, macula normală,periferie și vase OU.
curs
pacientul a fost admis la uihc pentru o lucrare de diagnostic, precum și tratament pentru insuficiență hepatică severă. Lucrările ulterioare au arătat înrăutățirea testelor funcției hepatice. El a avut ceruloplasmină serică normală și niveluri serice de cupru, dar a avut un cupru de urină crescut de 189 ng/ml (normal este de 12-80) și o excreție crescută de cupru de urină de 1522 mcg/zi (normal este de 3-35) după testul de excreție a penicilaminei. Biopsia sa hepatică a relevat o cuantificare a cuprului hepatic de 763 mcg de cupru pe gram de greutate uscată (normal este de 10-35 mcg/gm). datorită prezenței inelelor Kayser-Fleischer, excreției crescute a cuprului în urină după testul de excreție a penicilaminei și biopsiei hepatice diagnostice, pacientul a fost diagnosticat cu boala Wilson. Apoi a fost tratat cu penicilamină 250 mg de 4 ori pe zi și piridoxină 25 mg PO zilnic, ceea ce i-a îmbunătățit clearance-ul de cupru din urină. Cu toate acestea, el a dezvoltat encefalopatie semnificativă, o coagulopatie progresivă agravată cu hemoliză și anemie a bolii hepatice și ascite semnificative. Boala sa hepatică a continuat să progreseze pe parcursul celor două săptămâni de spital și a demonstrat un curs sub-fulminant. Hepatologii săi au considerat că boala va progresa probabil până la insuficiență hepatică și moarte probabilă. Transplantul de ficat nu a putut fi efectuat la UIHC din cauza istoricului său de consum de alcool și a rețelei sale slabe de sprijin. Apoi a fost transferat la un spital mai aproape de familia sa.
discuție: boala Wilson
boala Wilson, descrisă pentru prima dată în 1912 de Kinnear Wilson, este o boală autosomală recesivă rară de scădere a excreției biliare de cupru și a depunerii de cupru în organism, mai ales în ficat, creier, rinichi și cornee. Boala este cauzată de o mutație genetică a genei ATP7B de pe cromozomul 13 care codifică o ATPază transportoare de cupru legată de membrană găsită mai ales în ficat (Tanzi, 1993). Pacienții cu boala Wilson pot prezenta boli hepatice cronice, insuficiență hepatică fulminantă, insuficiență renală acută, anemie hemolitică sau boli neuropsihiatrice, cum ar fi tulburări de mișcare, tremor, necoordonare și modificări comportamentale (vezi Tabelul 1). Boala se poate prezenta la orice vârstă, dar este de obicei observată între deceniile 1 și 4 ale vieții (Schoen, 1990). Prevalența la nivel mondial a fost raportată a fi de 1 din 30.000 (Scheinberg, 1984). Lăsată netratată, această boală este letală. Tratamentele obișnuite cu penicilamină, trientină și/sau terapie cu zinc (tratamente de chelare a cuprului), precum și dietele cu conținut scăzut de cupru sunt eforturi de-a lungul vieții, transplantul hepatic salvând vieți în cazuri foarte avansate (Mak, 2008).
diagnosticarea bolii Wilson
boala Wilson este de obicei suspectată la pacienții tineri cu vârsta sub patruzeci de ani care au boli hepatice inexplicabile, boli neurologice comportamentale și/sau psihiatrice inexplicabile în contextul bolii hepatice, sau un istoric familial al bolii Wilson. Diagnosticul bolii Wilson se bazează adesea pe criteriile lui Sternlieb, unde un pacient trebuie să aibă cel puțin două dintre următoarele constatări: prezența inelelor Kayser-Fleischer, simptome neurologice tipice și/sau niveluri scăzute de ceruloplasmină (<0,20 g / L) (Sternlieb, 1990). Din păcate, aceste criterii sunt adesea îndeplinite numai atunci când un pacient prezintă o boală avansată și are de obicei manifestări neurologice și/sau psihiatrice. Pacienții cu boală precoce, numai boală hepatică sau boală asimptomatică sunt foarte dificil de diagnosticat.
nu există niciun test de laborator care să identifice în mod fiabil boala Wilson, dar o combinație de teste, inclusiv ceruloplasmina serică, cuprul fără ser și excreția urinară de cupru de 24 de ore, sunt utilizate împreună pentru a identifica anomalii ale metabolismului cuprului.
ceruloplasmina este o proteină produsă de hepatocite care leagă CUPRUL și îl livrează țesuturilor periferice. Când cuprul nu este disponibil, din cauza transportului transmembranar deficitar, pentru a fi încorporat în molecula de apoceruloplasmină ca în boala Wilson, apoceruloplasmina eliberată este metabolizată rapid, determinând nivelurile circulante ale ceruloplasminei să fie scăzute. Nivelul normal al ceruloplasminei din sânge este de 0,20 până la 0,40 g/L, cu toate acestea, această valoare nu se aplică pacienților pediatrici, pacienților gravide sau pacienților cu estrogen. Alte boli pot provoca niveluri scăzute, inclusiv malnutriție, sindrom nefrotic, aceruloplasminemie ereditară sau tulburări inflamatorii (deoarece este un reactant de fază acută). De asemenea, nivelurile de ceruloplasmină pot fi normale la pacienții cu boala Wilson, citate ca oriunde de la 35% -45% la pacienții cu prezentare hepatică și 60% dintre pacienții cu insuficiență hepatică fulminantă (Steindl, 1997). Prin urmare, la pacienții cu afecțiuni hepatice, un nivel normal de ceruloplasmină nu poate exclude boala Wilson și nici un nivel scăzut nu este suficient pentru a face un diagnostic al bolii Wilson.